











Cancer colorectal héréditaire sans polypose (HNPCC), aussi appelé Syndrome de Lynch, est une maladie génétique autosomique dominante associée à un risque élevé de cancer du côlon ainsi qu’à d’autres cancers, notamment le cancer de l’endomètre (le deuxième plus fréquent), l’ovaire, l’estomac, l’intestin grêle, les voies hépatobiliaires, les voies urinaires supérieures, le cerveau et la peau. Le risque accru de ces cancers est dû à des mutations héréditaires qui nuisent à la réparation des mésappariements de l’ADN. C’est un type de syndrome du cancer.
Symptômes du syndrome de Lynch
Risque de cancer
Risque à vie et âge moyen au diagnostic des cancers associés au syndrome de Lynch
| Type de cancer | Risque à vie (%) | Âge moyen au diagnostic (années) |
| Colorectal | 52-58 | 44-61 |
| Endomètre | 25-60 | 48-62 |
| Gastrique | 6-13 | 56 |
| Ovarien | 4-12 | 42,5 |
En plus des types de cancer trouvés dans le tableau ci-dessus, il est entendu que le syndrome de Lynch contribue également à un risque accru de cancer de l’intestin grêle, de cancer du pancréas, de cancer de l’uretère / du bassin rénal, de cancer des voies biliaires, de cancer du cerveau et de néoplasmes sébacés. Un risque accru de cancer de la prostate et de cancer du sein a également été associé au syndrome de Lynch, bien que cette relation ne soit pas entièrement comprise.
Les deux tiers des cancers du côlon surviennent dans le côlon proximal et les signes et symptômes courants comprennent le sang dans les selles, la diarrhée ou la constipation et la perte de poids involontaire. L’âge moyen du diagnostic de cancer colorectal est de 44 ans pour les membres de familles qui répondent aux critères d’Amsterdam. L’âge moyen du diagnostic du cancer de l’endomètre est d’environ 46 ans. Parmi les femmes atteintes de HNPCC qui ont à la fois un cancer du côlon et de l’endomètre, environ la moitié présentent d’abord un cancer de l’endomètre, ce qui fait du cancer de l’endomètre le cancer sentinelle le plus courant dans le syndrome de Lynch. Le symptôme le plus courant du cancer de l’endomètre est un saignement vaginal anormal. Dans HNPCC, l’âge moyen du diagnostic de cancer gastrique est de 56 ans, l’adénocarcinome de type intestinal étant la pathologie la plus fréquemment rapportée. Les cancers de l’ovaire associés au HNPCC ont un âge moyen de diagnostic de 42,5 ans; environ 30% sont diagnostiqués avant l’âge de 40 ans.
Une variation significative du taux de cancer a été trouvée en fonction de la mutation impliquée. Jusqu’à l’âge de 75 ans, les risques de cancer colorectal, de cancer de l’endomètre, de cancer de l’ovaire, du tube digestif supérieur (gastrique, duodénal, des voies biliaires ou pancréatique), des cancers des voies urinaires, du cancer de la prostate et des tumeurs cérébrales étaient les suivants: pour les mutations MLH1, le risque était respectivement de 46%, 43%, 10%, 21%, 8%, 17% et 1%; pour les mutations MSH2, les risques étaient respectivement de 57%, 17%, 10%, 25%, 32% et 5%; pour les mutations MSH6, les risques étaient respectivement de 15%, 46%, 13%, 7%, 11%, 18% et 1%.
| Gène | Risque de cancer de l’ovaire | Risque de cancer de l’endomètre |
|---|---|---|
| MLH1 | 4 à 24% | 25 à 60% |
| MSH2 / EPCAM | 4 à 24% | 25 à 60% |
| MSH6 | 1 à 11% | 16 à 26% |
| PMS2 | 6% (risque combiné) | 15% |
La génétique

HNPCC est hérité de manière autosomique dominante. La caractéristique de HNPCC est la réparation défectueuse des mésappariements d’ADN, ce qui entraîne un taux élevé de modifications d’un seul nucléotide et une instabilité des microsatellites, également connue sous le nom de MSI-H (le H est «élevé»). Le MSI est identifiable dans les échantillons de cancer du laboratoire de pathologie. La plupart des cas entraînent des modifications de la longueur des répétitions dinucléotidiques des nucléobases cytosine et adénine (séquence: CACACACACA…).
Les 4 principaux gènes impliqués dans HNPCC codent normalement pour des protéines qui forment des dimères pour fonctionner:
- La protéine MLH1 se dimérise avec la protéine PMS2 pour former MutLα, qui coordonne la liaison d’autres protéines impliquées dans la réparation des mésappariements comme l’ADN hélicase, la protéine de liaison à l’ADN simple brin (RPA) et les ADN polymérases.
- La protéine MSH2 se dimérise avec la protéine MSH6, qui identifie les mésappariements via un modèle de pince coulissante, une protéine permettant de détecter les erreurs.
La dégradation de l’un ou l’autre gène pour le dimère de protéine altère la fonction de la protéine. Ces 4 gènes sont impliqués dans la correction des erreurs (réparation des mésappariements), de sorte que le dysfonctionnement des gènes peut conduire à l’incapacité de corriger les erreurs de réplication de l’ADN et provoquer le HNPCC. Le HNPCC est connu pour être associé à d’autres mutations dans les gènes impliqués dans la voie de réparation des mésappariements d’ADN:
| Nom OMIM | Gènes impliqués dans HNPCC | Fréquence des mutations dans les familles HNPCC | Lieu | Première publication |
|---|---|---|---|---|
| HNPCC1 (120435) | MSH2 / EPCAM | environ 60% | 2p22 | Fishel 1993 |
| HNPCC2 (609310) | MLH1 | environ 30% | 3p21 | Papadopoulos 1994 |
| HNPCC5 | MSH6 | 7 à 10% | 2p16 | Miyaki 1997 |
| HNPCC4 | PMS2 | relativement peu fréquent | 7p22 | Nicolaides 1994 |
| HNPCC3 | PMS1 | rapport de cas | 2q31-q33 | Nicolaides 1994 |
| HNPCC6 | TGFBR2 | rapport de cas | 3p22 | |
| HNPCC7 | MLH3 | contesté | 14q24.3 |
La plupart des personnes atteintes de HNPCC héritent de la maladie d’un parent. Cependant, en raison d’une pénétrance incomplète, de l’âge variable du diagnostic de cancer, de la réduction du risque de cancer ou de la mort prématurée, toutes les personnes atteintes d’une mutation du gène HNPCC n’ont pas un parent qui avait un cancer. Certaines personnes développent le HNPCC de-novo dans une nouvelle génération, sans hériter du gène. Ces personnes ne sont souvent identifiées qu’après avoir développé un cancer du côlon au début de la vie. Les parents atteints de HNPCC ont 50% de chances de transmettre la mutation génétique à chaque enfant. Il est également important de noter qu’une mutation délétère dans l’un des gènes MMR seul n’est pas suffisante pour provoquer le cancer, mais que d’autres mutations dans d’autres gènes suppresseurs de tumeur doivent se produire.
Diagnostic du syndrome de Lynch
Un diagnostic de syndrome de Lynch est appliqué aux personnes ayant une mutation de l’ADN germinatif dans l’un des gènes MMR (MLH1, MSH2, MSH6 et PMS2) ou le gène EPCAM, identifié par des tests génétiques. Les candidats aux tests génétiques de la lignée germinale peuvent être identifiés par des critères cliniques tels que les critères cliniques d’Amsterdam et les directives Bethesda, ou par l’analyse des tumeurs par immunohistochimie (IHC) ou les tests d’instabilité des microsatellites (MSI). Aux États-Unis, les sociétés professionnelles recommandent de tester chaque cancer du côlon pour MSI ou IHC comme dépistage du syndrome de Lynch, mais cela n’est pas toujours effectué en raison des limites de coût et de ressources. Les tests génétiques sont disponibles dans le commerce et consistent en un test sanguin.
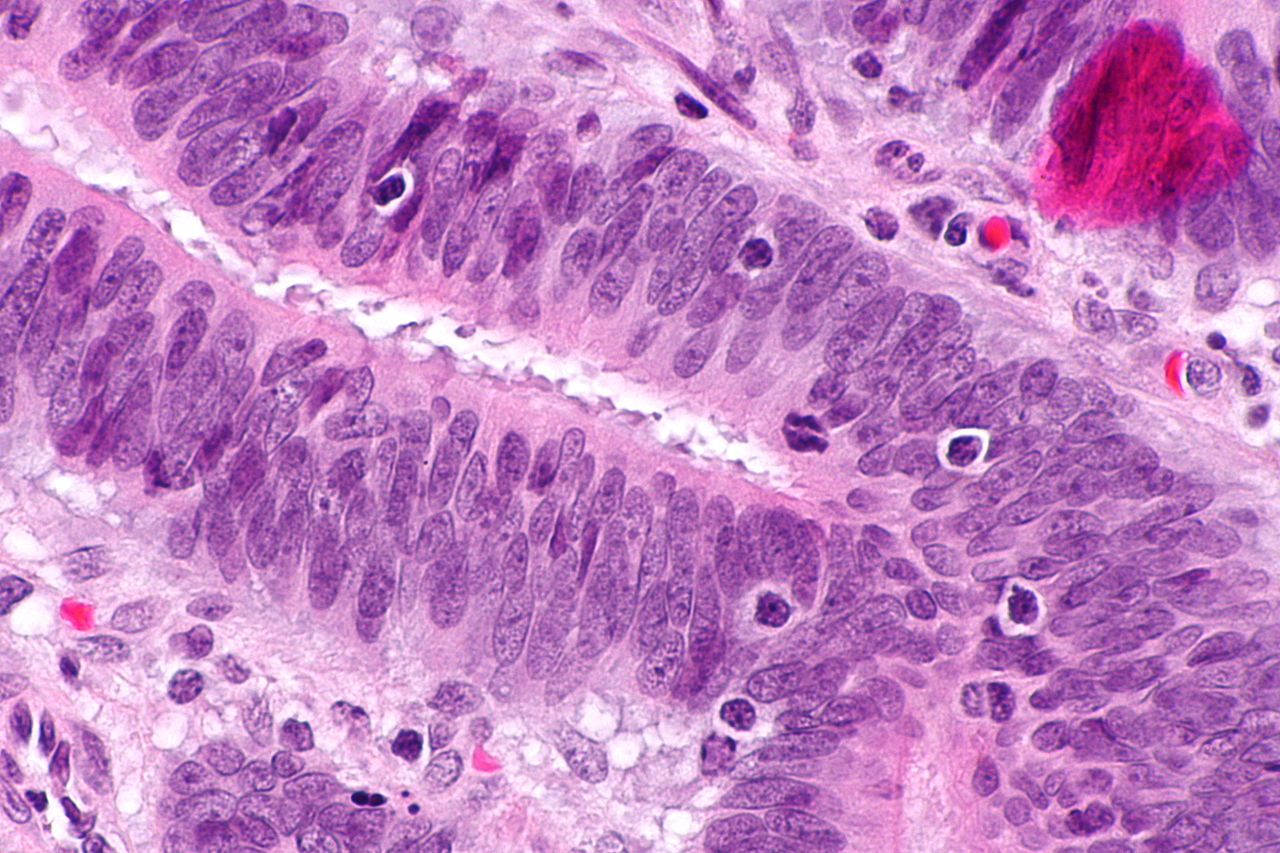
Immunohistochimie
L’immunohistochimie (IHC) est une méthode qui peut être utilisée pour détecter l’expression de la protéine ROR (mismatch repair) anormale dans les tumeurs associées au syndrome de Lynch. Bien qu’il ne soit pas un diagnostic du syndrome de Lynch, il peut jouer un rôle dans l’identification des personnes qui devraient subir des tests germinatifs. Deux méthodes de mise en œuvre des tests IHC comprennent les tests basés sur l’âge et les tests universels pour toutes les personnes. À l’heure actuelle, il n’y a pas d’accord général sur la méthode de dépistage à utiliser. Des tests d’IHC basés sur l’âge ont été suggérés en partie en raison d’analyses coûts-avantages, tandis que les tests universels pour toutes les personnes atteintes de cancer colorectal garantissent que les personnes atteintes du syndrome de Lynch ne sont pas manquées. Pour faire face aux coûts, les chercheurs tentent de prédire le MSI ou l’IHC directement à partir de l’apparence de la tumeur au microscope, sans effectuer de test moléculaire.
Instabilité des microsatellites
Les mutations dans les systèmes de réparation des mésappariements d’ADN peuvent entraîner des difficultés à transmettre des régions au sein de l’ADN qui contiennent des motifs répétitifs de deux ou trois nucléotides (microsatellites), également connus sous le nom d’instabilité des microsatellites (MSI). Le MSI est identifié par extraction d’ADN à la fois d’un échantillon de tissu tumoral et d’un échantillon de tissu normal, suivie d’une analyse par PCR des régions microsatellites. L’analyse MSI peut être utilisée pour identifier les personnes susceptibles d’être atteintes du syndrome de Lynch et les diriger vers des tests supplémentaires.
Classification
Trois grands groupes de cancers MSI-H (instabilité microsatellitaire – MSI) peuvent être reconnus par des critères histopathologiques:
- cancers mal différenciés du côté droit
- cancers mucineux du côté droit
- adénocarcinomes à n’importe quel endroit montrant n’importe quel niveau mesurable de lymphocytes intraépithéliaux (TIL)
Les critères histopathologiques ne sont pas suffisamment sensibles pour détecter le MSI à partir de l’histologie, mais les chercheurs tentent d’utiliser l’intelligence artificielle pour prédire le MSI à partir de l’histologie.
De plus, le HNPCC peut être divisé en syndrome de Lynch I (cancer du côlon familial) et syndrome de Lynch II (HNPCC associé à d’autres cancers du tractus gastro-intestinal ou du système reproducteur).
La prévention
Test de dépistage
Le conseil génétique et les tests génétiques sont recommandés pour les familles qui répondent aux critères d’Amsterdam, de préférence avant l’apparition du cancer du côlon.
Cancer du colon
Les coloscopies sont recommandées comme méthode préventive de surveillance pour les personnes atteintes du syndrome de Lynch ou de gènes associés au LS. Plus précisément, il est recommandé que les coloscopies commencent entre 20 et 25 ans pour les porteurs de mutations MLH1 et MSH2 et 35 ans pour les porteurs de mutations MSH6 et PMS2. Une surveillance coloscopique doit alors être effectuée à un intervalle de 1 à 2 ans pour les patients atteints du syndrome de Lynch.
Cancer de l’endomètre / de l’ovaire
Une échographie transvaginale avec ou sans biopsie de l’endomètre est recommandée chaque année pour le dépistage du cancer de l’ovaire et de l’endomètre. Pour les femmes atteintes du syndrome de Lynch, un test sanguin annuel CA-125 peut être utilisé pour dépister le cancer de l’ovaire, mais il existe des données limitées sur l’efficacité de ce test pour réduire la mortalité.
Autres cancers
Il existe également des stratégies pour détecter précocement d’autres cancers ou réduire les chances de les développer dont les personnes atteintes du syndrome de Lynch peuvent discuter avec leur médecin, mais leur efficacité n’est pas claire. Ces options comprennent:
- Endoscopies supérieures pour détecter le cancer de l’estomac et de l’intestin grêle tous les 3 à 5 ans, à partir de 30 ans au plus tôt (de préférence dans un cadre de recherche)
- Analyse d’urine annuelle pour détecter le cancer de la vessie, à partir de 30 ans au plus tôt (de préférence dans un cadre de recherche)
- Examens physiques et neurologiques annuels pour détecter le cancer du système nerveux central (cerveau ou moelle épinière), à partir de 25 ans au plus tôt
Critères d’Amsterdam
Voici les critères d’Amsterdam pour l’identification des candidats à haut risque pour les tests de génétique moléculaire:
Critères d’Amsterdam I (tous les points doivent être remplis):
- Trois membres de la famille ou plus avec un diagnostic confirmé de cancer colorectal, dont l’un est un parent au premier degré (parent, enfant, frère ou sœur) des deux autres
- Deux générations successives affectées
- Un ou plusieurs cancers du côlon diagnostiqués avant l’âge de 50 ans
- La polypose adénomateuse familiale (FAP) a été exclue
Les critères d’Amsterdam II ont été développés en 1999 et ont amélioré la sensibilité diagnostique du syndrome de Lynch en incluant les cancers de l’endomètre, de l’intestin grêle, de l’uretère et du bassin rénal.
Critères d’Amsterdam II (tous les points doivent être remplis):
- Trois membres de la famille ou plus atteints de cancers liés au HNPCC, dont l’un est un parent au premier degré des deux autres
- Deux générations successives affectées
- Un ou plusieurs cancers liés au HNPCC diagnostiqués avant l’âge de 50 ans
- La polypose adénomateuse familiale (FAP) a été exclue
Opération
Une hystérectomie prophylactique et une salpingo-ovariectomie (ablation de l’utérus, des trompes de Fallope et des ovaires pour empêcher le cancer de se développer) peuvent être pratiquées avant le développement d’un cancer de l’ovaire ou de l’endomètre.
Traitement du syndrome de Lynch
La chirurgie demeure la thérapie de première ligne pour le HNPCC. Les patients atteints du syndrome de Lynch qui développent un cancer colorectal peuvent être traités soit par une colectomie partielle, soit par une colectomie totale avec anastomose iléo-rectale. En raison du risque accru de cancer colorectal après une colectomie partielle et d’une qualité de vie similaire après les deux chirurgies, une colectomie totale peut être un traitement préféré pour le HNPCC, en particulier chez les patients plus jeunes.
Il existe une controverse en cours sur les avantages des thérapies adjuvantes à base de 5-fluorouracile pour les tumeurs colorectales liées au HNPCC, en particulier celles des stades I et II.
- La thérapie par anticorps anti-PD-1 peut être efficace.
Le blocage des points de contrôle avec un traitement anti-PD-1 est maintenant le traitement de première intention préféré pour le cancer colorectal avancé à instabilité des microsatellites – haute.
.






:max_bytes(150000):strip_icc()/stages-of-alzheimers-dementia-4589632-01-15e9165202c140e8875d28ee2aeae092.png)

Discussion about this post